




Константин Валерьевич Балакин, д. х. н., проф. Московского физико-технического института.
Email: balakin.kv@mipt.ru
Андрей Александрович Иващенко, д. т. н., зав. кафедрой инновационной фармацевтики, медицинской техники и биотехнологий Московского физико-технического института, председатель совета директоров группы компаний «ХимРар», руководитель Комитета по инновационной фармацевтике ассоциации «Национальные чемпионы».
В 2012 году Управление по санитарному надзору за качеством пищевых продуктов и медикаментов США (FDA) ввело статус «прорывная терапия» для обозначения специальной категории инновационных лекарственных средств. Целью этой программы является упрощение и ускорение разработки лекарственных препаратов нового поколения для лечения серьезных заболеваний, не имевших до этого специфических средств терапии. С тех пор более 130 новых лекарственных препаратов получили в США статус «прорывных». В чем состоят ключевые особенности этой категории лекарств? Какова их реальная инновационность? Насколько они безопасны и эффективны? Что думают о «прорывных» лекарствах врачи, пациенты и инвесторы? Какие уроки для Российской Федерации могут быть вынесены из анализа этой программы? Данная статья посвящена этим и другим сопутствующим вопросам.
В 2012 году Конгресс США ввел новую процедуру рассмотрения лекарственных препаратов для FDA – программу «прорывная терапия» (англ. breakthrough therapy). Этим статусом может быть наделен «препарат, предназначенный для лечения серьезного заболевания, если предварительные клинические данные указывают на то, что этот препарат может продемонстрировать существенное улучшение клинически значимой конечной точки (точек) по сравнению с имеющимися методами лечения» [1]. В качестве обоснования разработчики новой процедуры приводили данные о том, что эффективность современных лекарственных препаратов возможно достоверно наблюдать уже во второй фазе клинических исследований (КИ), и поэтому масштабные рандомизированные КИ третьей фазы не являются совершенно необходимыми для получения разрешения регулирующих органов. Более того, ситуацию, когда пациенты с тяжкими и порою неизлечимыми заболеваниями годами вынуждены ждать результаты длительных исследований третьей фазы препаратов, которые не имеют альтернатив и при этом показали убедительные результаты по эффективности и безопасности на предшествующих этапах, следует считать неэтичной.
Ключевой особенностью новой регуляторной процедуры по сравнению со стандартной процедурой является отмена необходимости в третьей фазе КИ (рис. 1), хотя и с усилением постмаркетингового надзора (четвертая фаза КИ). Программа также предусматривает более ранние (уже в ходе первой фазы КИ) и более частые контакты разработчика с ведущими экспертами FDA, чтобы направить процесс разработки лекарств по оптимальному исследовательскому и регуляторному треку. Одним из ключевых условий для получения статуса «прорывная терапия» является очень значимая предполагаемая польза от нового препарата. В отличие от других программ ускоренного рассмотрения FDA, эта польза может быть оценена на основании определенной суррогатной конечной точки, которая может предсказать клинический исход обоснованных биомаркеров или улучшенного профиля безопасности и эффективности относительно стандарта лечения.
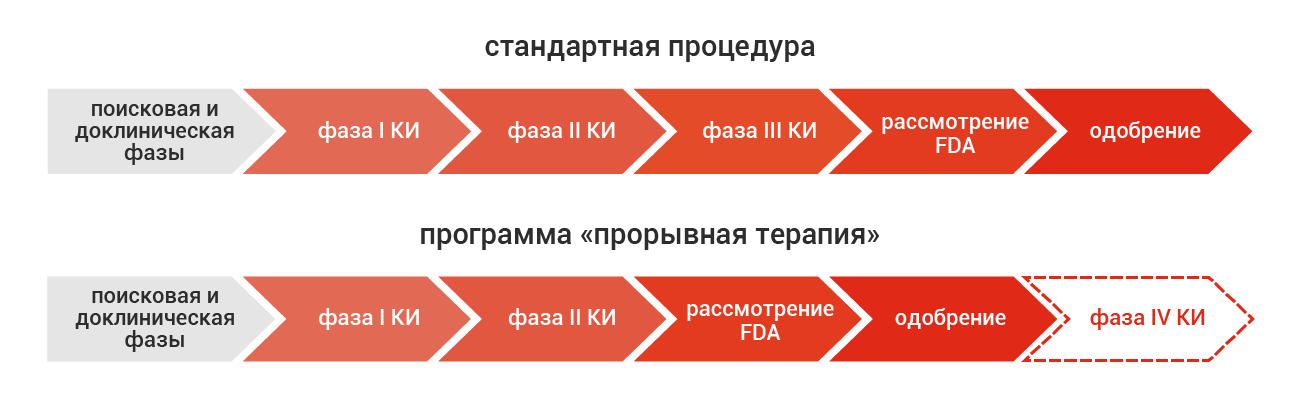 Рис. 1. Сравнение процедур стандартного рассмотрения и программы «прорывная терапия» в FDA США.
Рис. 1. Сравнение процедур стандартного рассмотрения и программы «прорывная терапия» в FDA США.
После введения в 2012 году новая программа быстро набрала популярность. Так, до конца 2022 года FDA получило в общей сложности 1289 заявок на «прорывную терапию»; более трети заявок были удовлетворены, что в итоге привело к одобрению 125 новых «прорывных» препаратов [1] (рис. 2); в 2023 году этот список пополнился еще девятью препаратами.
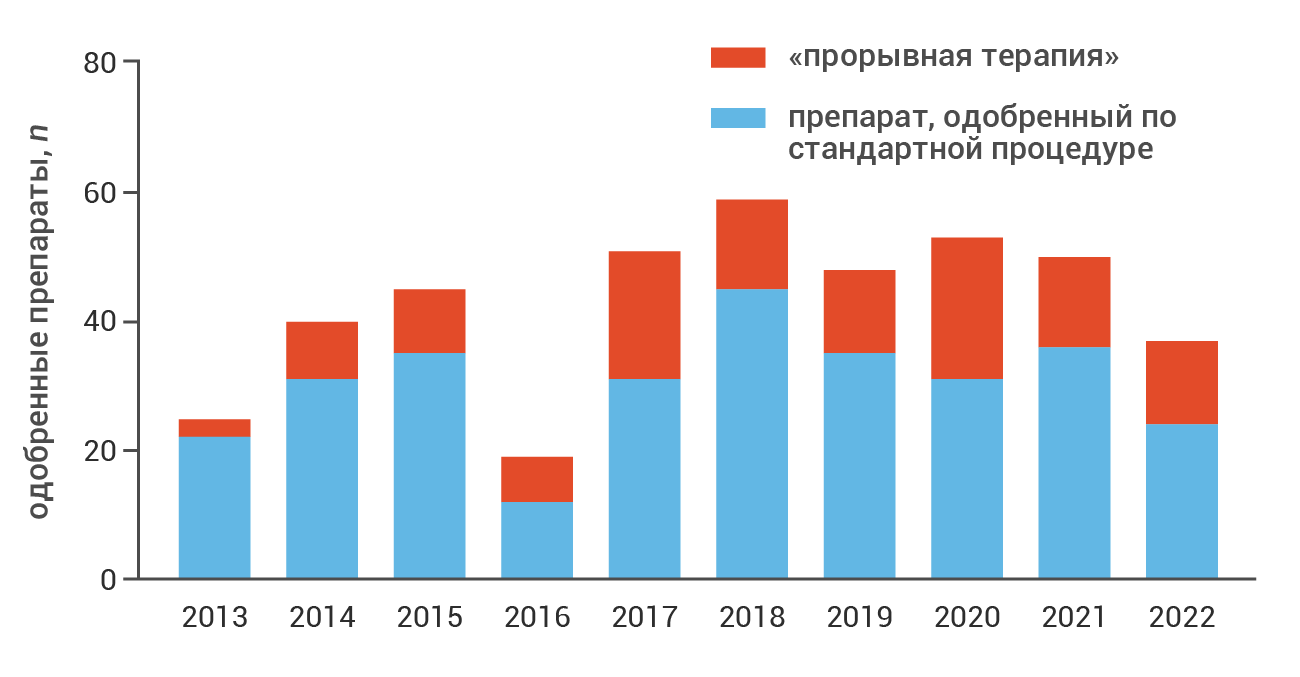 Рис. 2. Динамика регистрации «прорывных» препаратов в США в сравнении с одобрениями по стандартной процедуре [1].
Рис. 2. Динамика регистрации «прорывных» препаратов в США в сравнении с одобрениями по стандартной процедуре [1].
Насколько безопасны и эффективны «прорывные» препараты?
С самого начала новая программа, отменяющая необходимость в проведении третьей фазы КИ, вызвала серьезную полемику в среде законодателей и медицинских специалистов по поводу безопасности и эффективности новых препаратов. В целом ряде исследований было отмечено, что «прорывные» препараты часто утверждаются на основании нерандомизированных, однокогортных исследований, что может приводить к более высоким рискам серьезных нежелательных явлений. Тем не менее, хотя вопрос о влиянии этого статуса на безопасность остается открытым, к настоящему времени не было обнаружено достоверных доказательств негативного влияния [1], [2].
Также по-прежнему ведутся споры об эффективности «прорывных» препаратов. В ряде работ не было выявлено существенной разницы в показателях клинической эффективности между препаратами со статусом «прорывных» или без него [2], [3]. Однако в крупнейшем и самом свежем на сегодняшний день исследовании 355 «прорывных» и «непрорывных» противоопухолевых средств, одобренных FDA с 2012 по 2022 год, было показано, что «прорывные» препараты ассоциировались с меньшей вероятностью смерти (отношение рисков: 0,69 против 0,74, p=0,031) и обеспечивали значительно большее улучшение медианы общей выживаемости (4,8 против 3,2 месяца, p=0,004) [4]. Было также показано, что вероятность того, что «прорывные» препараты будут иметь высокую терапевтическую ценность, в четыре раза выше [5]. То есть имеющиеся на сегодня данные позволяют говорить о положительной корреляции между «прорывным» статусом и эффективностью препаратов.
Насколько инновационны «прорывные» препараты?
Не вызывает сомнений, что «прорывные» препараты – это наиболее инновационная группа новых регистрируемых лекарств в США. Среди них высока доля структур типа «первые в классе»; кроме того, такие препараты чаще лечат новые заболевания, действуют по инновационному механизму, а также чаще принадлежат к новым типам лекарств.
В качестве примеров действительно уникальных и прорывных (без кавычек!) достижений можно привести такие препараты, как ленакапавир (средство терапии инфекции ВИЧ, действующее по механизму ингибирования вирусного капсида) и соторасиб (противоопухолевый ингибитор мутантной формы ГТФазы KRas G12C) (рис. 3). Не будет преувеличенным утверждение, что подобные препараты открывают даже не новые страницы, а новые главы в своих терапевтических областях.
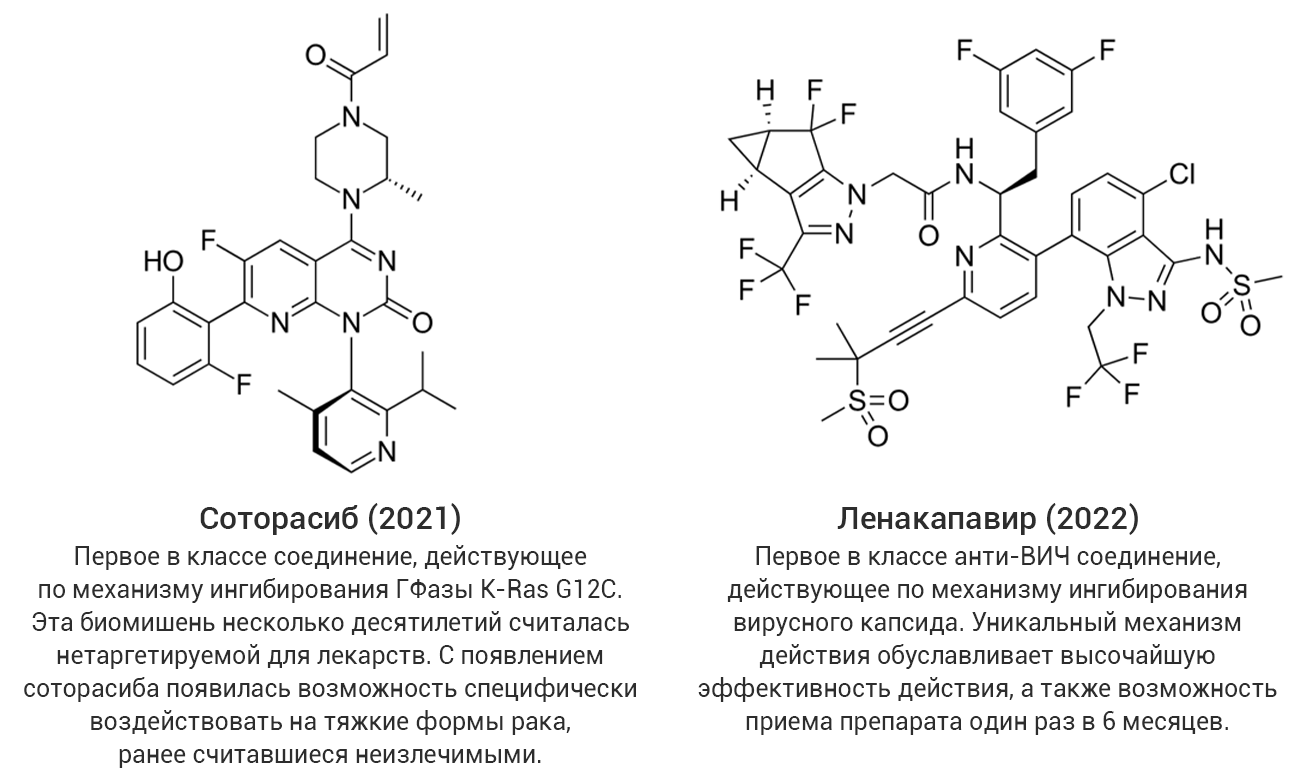 Рис. 3. Примеры уникальных высокоинновационных соединений со статусом «прорывных».
Рис. 3. Примеры уникальных высокоинновационных соединений со статусом «прорывных».
Однако даже среди «прорывных» молекул немало таких, которые представляют собой близкие структурные аналоги уже известных лекарственных соединений, в том числе активно применяющихся на практике. С точки зрения медицинской химии такие новые лекарства разработаны с использованием стратегии дизайна на основе аналога (англ. analog-based design), широко применяемой в современной научно-индустриальной практике. В качестве примера на рис. 4 (а-г) показаны четыре пары соединений, каждая из которых представляет собой прототип-предшественник (слева) и его более поздний структурный аналог со статусом «прорывного» (справа). В двух последних случаях показаны самые свежие представители данной категории лекарственных средств, одобренные в 2024 году, – ресметиром и апроситентан, являющиеся аналогами более ранних предшественников.
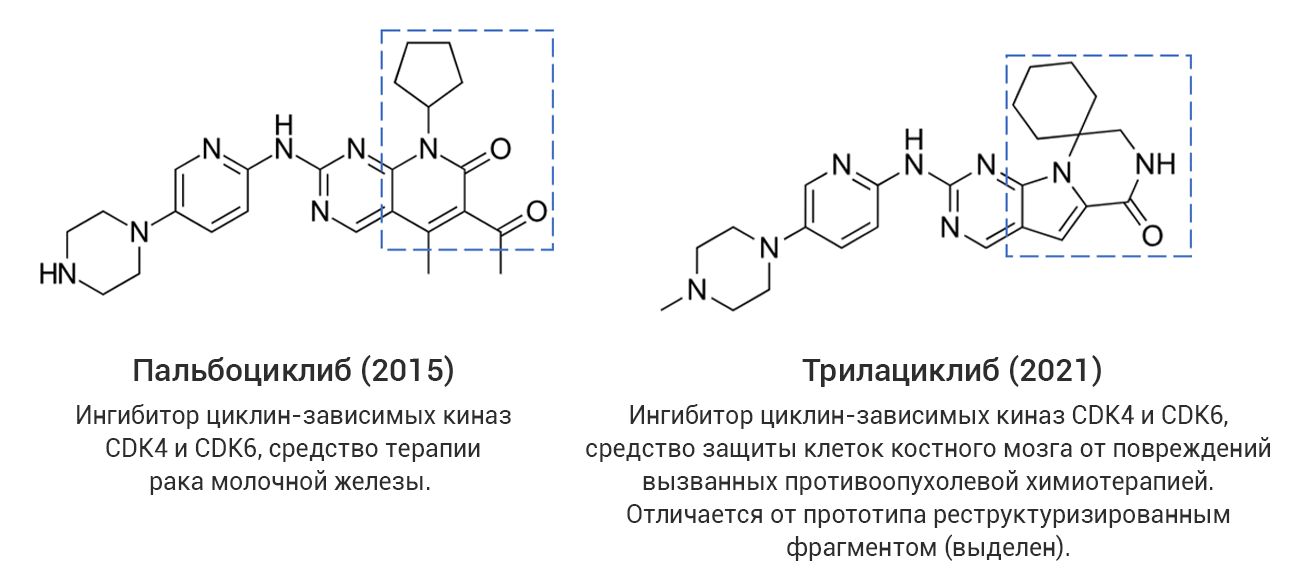
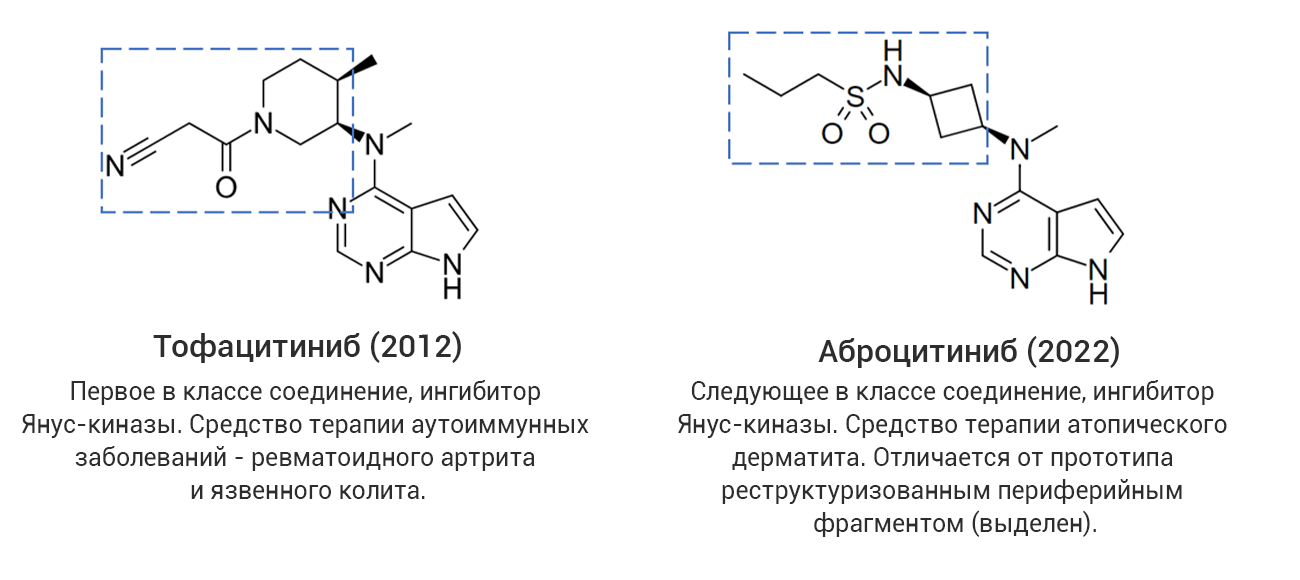
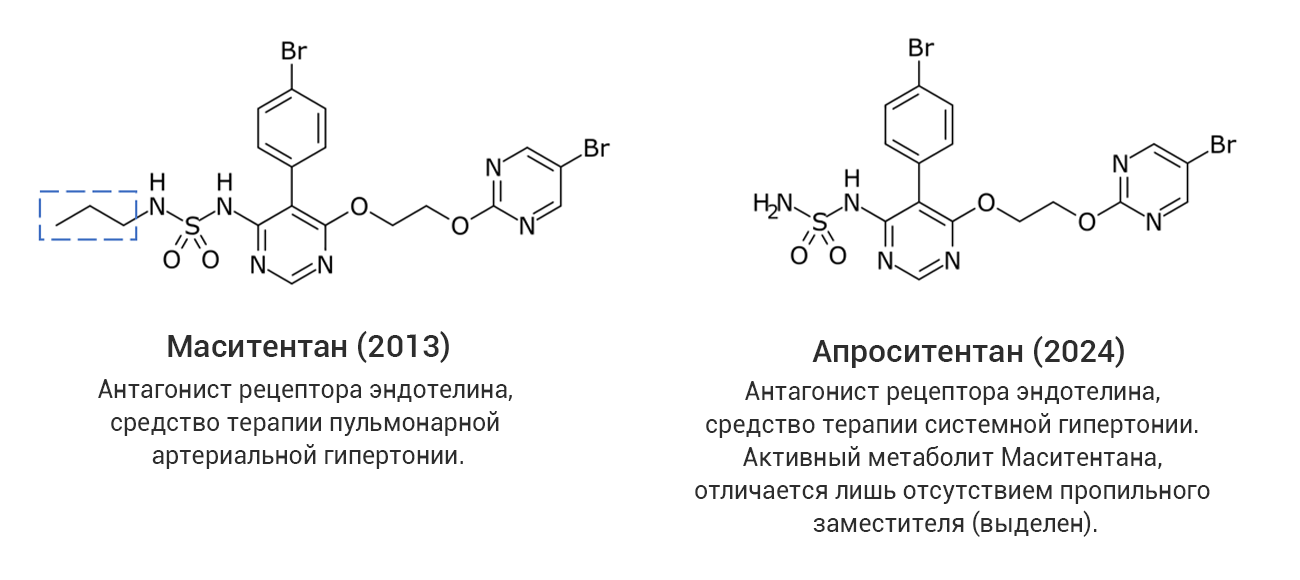
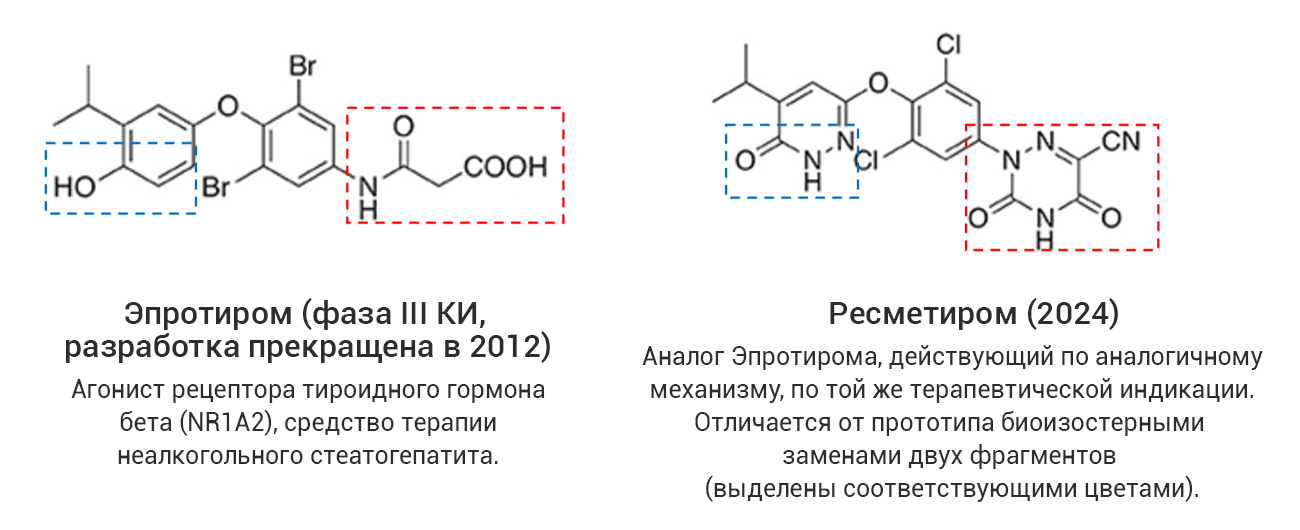 Рис. 4. Примеры лекарственных аналогов среди «прорывных» препаратов.
Рис. 4. Примеры лекарственных аналогов среди «прорывных» препаратов.
Из этих примеров следует, что статус «прорывных» могут получить не обязательно высокоинновационные с точки зрения современной медицинской химии соединения. В связи с этим следует обратить внимание, что официальное определение FDA не содержит явного указания на инновационность или высокотехнологичность «прорывного» препарата.
Что думают о «прорывных» лекарствах врачи, пациенты и инвесторы?
Несмотря на отсутствие в большинстве случаев убедительных доказательств того, что «прорывные» препараты более безопасны или эффективны, пациенты и врачи обычно связывают с этим статусом весьма оптимистичные ожидания. В соответствии с недавним исследованием, повышенная доля пациентов верит в то, что «прорывной» препарат более эффективен по сравнению с функциональным аналогом без этого статуса (11% против 25%, p=0,001) [6]. При выборе между двумя препаратами с одинаковой безопасностью, эффективностью, доказательной базой и стоимостью 94% врачей предпочитают назначить препарат со статусом «прорывного» [7]. Как следствие отмеченного оптимизма потребителей и врачей, ежемесячные расходы на лечение «прорывными» препаратами примерно на $16 000 выше по сравнению с «непрорывными» ($38 971 против $22 591, p=0,0592) [3], [4], что, очевидно, является положительным сигналом для инвесторов.
В табл. 1 представлено влияние статуса «прорывная терапия» на некоторые значимые медицинские и рыночные аспекты, относящиеся к новому лекарственному препарату.
Табл. 1. Влияние прохождения по категории «прорывная терапия» на некоторые значимые аспекты, относящиеся к лекарственному препарату.
| Аспект | Влияние статуса «прорывная терапия» |
| Безопасность | Потенциально отрицательное, но статистически незначимое в исследованных случаях |
| Эффективность | Положительное, хотя во многих случаях статистически незначимое |
| Медицинская доказательная база | Уменьшение клинических доказательных данных в связи с отсутствием третьей фазы КИ |
| Инновационность | В целом положительное, но без формальных регуляторных требований; немалая доля «прорывных» препаратов являются аналогами известных структур |
| Инвестиционная привлекательность | Положительное |
| Длительность процесса разработки | Значимое уменьшение сроков разработки и выведения на рынок |
| Стоимость препарата | Значимое увеличение рыночной стоимости |
Ключевые уроки для нашей страны
Мировой опыт свидетельствует о том, что определенная часть новых лекарственных средств, предназначенных для терапии тяжких заболеваний, может получить достаточно убедительные доказательства эффективности и безопасности уже по итогам второй фазы КИ. Для таких препаратов имеет смысл законодательно обеспечить ускоренное рассмотрение и регистрацию, что отвечает интересам как пациентов, так и организаций-разработчиков, поскольку сокращает время появления продукта на рынке и снижает расходы на разработку. Обратной стороной медали для пациентов, как правило, является более высокая стоимость препаратов, и в таких случаях нелишними могут быть меры государственного регулирования цены.
Наиболее систематический и в целом успешный опыт развития программы «прорывной терапии» накоплен с 2012 года в США, хотя сходные программы имеются также в ЕС [8] и КНР [9]. Целый ряд «прорывных» препаратов, по версии FDA, могут быть востребованы российским здравоохранением для лечения серьезных социально значимых патологий или послужить в качестве прототипов для разработки их улучшенных модификаций типа «следующие в классе».
Можно предположить, что наиболее удачные решения из практики ведущих национальных и региональных регуляторных ведомств могут быть с успехом перенесены на российскую почву, тем более что успешный опыт подобного рода в нашей стране уже имеется. Так, Постановление Правительства Российской Федерации от 3 апреля 2020 года № 441 «Об особенностях обращения лекарственных препаратов для медицинского применения…» [10] позволило в предельно короткие сроки обеспечить население России эффективными антиковидными средствами собственной разработки, такими как «Авифавир» (ГК «ХимРар»), «Левилимаб» (Biocad), вакцина «Cпутник V» (НИЦЭМ им. Гамалеи) и другие. В современной драматичной геополитической обстановке снижение регуляторных барьеров на пути к рыночному внедрению перспективных отечественных инновационных лекарственных средств может содействовать преодолению технологической зависимости от западной фарминдустрии и достижению технологического суверенитета в сфере лекарственного обеспечения.
Литература
[1] Michaeli D.T. et al. Special FDA designations for drug development: orphan, fast track, accelerated approval, priority review, and breakthrough therapy. Eur. J. Health Econ. 2023. https://doi.org/10.1007/s10198-023-01639-x.
[2] Hwang T.J. et al. Efficacy, safety, and regulatory approval of food and drug administration-designated breakthrough and nonbreakthrough cancer medicines. J. Clin. Oncol. 36, 1805–1812 (2018).
[3] Molto C. et al. Clinical benefit and cost of breakthrough cancer drugs approved by the US Food and Drug Administration. Cancer 126, 4390–4399 (2020).
[4] Michaeli D.T. et al. Breakthrough Therapy Cancer Drugs and Indications with FDA Approval: Development Time, Innovation, Trials, Clinical Benefit, Epidemiology, and Price. J. Natl. Compr. Canc. Netw. 1-9 (2024).
[5] Hwang T.J. et al. Association between FDA and EMA expedited approval programs and therapeutic value of new medicines: retrospective cohort study. The BMJ 371, m3434 (2020).
[6] Chandra A. et al. Regulatory Incentives for Innovation: The FDA’s Breakthrough Therapy Designation. https://doi.org/10.2139/ssrn.4296623 (2022).
[7] Krishnamurti T. et al. A randomized trial testing US food and drug administration “breakthrough” language. JAMA Intern. Med. 175, 1856–1858 (2015).
[8] EMA: Accelerated assessment. In: Eur. Med. Agency. https://www.ema.europa.eu/en/human-regulatory/marketing-authorisation/accelerated-assessment (2018). Ссылка по состоянию на 11.06.2024.
[9] NMPA Issues the Announcement on Three Documents Including the Working Procedures for Review of Breakthrough Therapy Drugs (Interim). https://english.nmpa.gov.cn/2020-07/07/c_538688.htm (2020). Ссылка по состоянию на 11.06.2024.
[10] Постановление Правительства Российской Федерации от 03.04.2020 № 441. http://publication.pravo.gov.ru/Document/View/0001202004060038. Ссылка по состоянию на 11.06.2024.
